
In recent decades, bioinformatics has become an indispensable branch of modern scientific research, experienced an explosion in financial support, application development and data collection. Growth dataset emerging from research laboratories, industry, health sector, etc., increase the level of demand in computing power and storage.
Biological data processing, large-scale datasets, often require the use of High Performance Computing (HPC) resources, especially when dealing with certain types of data omics, such as genomic and metagenomic the data. resources such as computing not only require substantial investment, but they also involve high maintenance costs.
More importantly, to maintain good returns on investment, specialized training should be put in place to ensure that waste is minimized. Moreover, given that bioinformatics is a field that is highly interdisciplinary in which several other domains intersect (such as biology, chemistry, physics and computer science), researchers from the areas also require training in bioinformatics in HPC, in order to fully utilize the centers supercomputer.
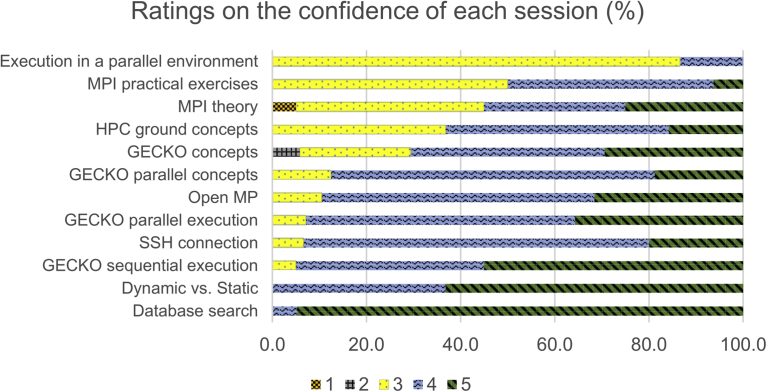
In this document, we describe our experience in the training of researchers from several different disciplines in HPC, as applied to bioinformatics in the context of Europe’s leading bioinformatics platform ELIXIR, and analyze both the content and outcome of the course.
Functional classification of protein structures by local structure matching in graph representation.
As a result of initiatives of high-throughput protein structure, over 14,400 protein structures have been solved by the Structural Genomics (SG) centers and participating research groups. While the totality of data SG is an outstanding contribution to genomics and structural biology, functional reliable information for this protein is generally lacking. better functional predictions for proteins SG will add great value to structural information has been obtained.
Our method described herein, Graphic Representation of the active site for the Prediction Function (GRASP-Func), quickly and accurately predict biochemical function of proteins by representing residue predicted at the local site is active as a graph rather than in Cartesian coordinates.
We compared the methods of GRASP-Func to our method previously reported, a structural block Local Site Activities (SALSA), using ribulose phosphate Binding Barrel (RPBB), 6-Hairpin Glycosidase (6-HG), and concanavalin A-like lectin / glucanase (CAL / G) superfamilies as test cases. In each superfamilies, SALSA and faster methods of GRASP-Func produce the correct classification similar to that previously characterized proteins, provide a benchmark validated for the new method. In addition, we analyzed protein and SG using our SALSA-Func GRASP method for predicting the function.
Forty-one SG at RPBB protein superfamily, nine in 6-SG protein superfamily HG and SG protein in the CAL / G superfamily successfully classified into one functional families within the superfamily each with both methods. , Faster, enhanced validated computational method can produce a more reliable prediction of the functions that can be used for various applications by the public.