
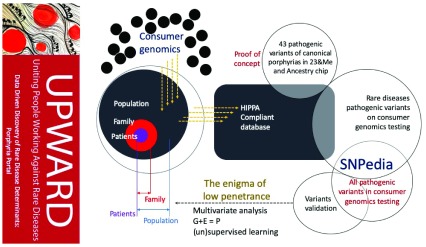
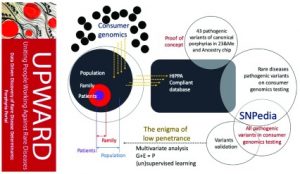
Background: The scientific basic and clinical research at the University of South Florida (USF) intersected to support a multifaceted approach around a common goal on rare diseases related to iron.
We proposed a modified version of the National Center (NCBI) Hackathon information biotechnology model to take full advantage of local expertise in the construction of “Iron Hack,” a rare hackathon focused on diseases. As the collaborative, problem solving of hackathons tends to attract participants from very different backgrounds, the organizers have hosted a symposium on rare diseases related to iron, especially porphyria and Friedreich’s ataxia, drawn to the general public .
Methods: The hackathon was structured to start each day with presentations by expert clinicians, genetic counselors, researchers focused on molecular and cellular biology, public / global health health, genetics / genomics, computational biology , bioinformatics, biomolecular science, bioengineering and computer science, as well as guest speakers from the American Foundation porphyria (APF) and the Friedreich’s Ataxia research Alliance (FARA) to inform participants on the human impact of these diseases.
Results: Because of this Hackathon, we have developed resources that are relevant not only to these specific models-diseases, but also to other rare diseases and problems of bioinformatics in general. In the two and a half days, the participants’ Iron Hack »successfully integrated collaborative projects to visualize the data, building databases to improve the diagnosis of rare diseases, and to study the legacy rare disease.
Conclusions: The purpose of this manuscript is to demonstrate the usefulness of a hackathon model to generate prototypes of generalized tools for a given disease and train clinicians and science interact effectively.
DataPackageR: Reproducible data preprocessing, standardization and sharing using R/Bioconductor for collaborative data analysis.
A central tenet of the study was that the reproducible scientific results published along with the underlying data and the software code needed to reproduce and verify the findings. A number of tools and software has been released which facilitates such as work-flows and scientific journals are increasingly demanding that the code and the primary data made available by the publication.
There is little practical advice on the implementation of reproducible research work for a large flow ‘omics’ or the biological systems data sets used by the analyst team working together. In such cases, it is important to ensure all analysts are using the same version of a set of data for their analysis. However, instantiating relational databases and standard operating procedures could be severe, with high “startup” costs and non-compliance with the procedure when they deviate substantially from a regular analyst workflow.
Ideally reproduced workflow research should fit naturally into the existing individual work-flow, with minimal disruption. Here, we provide an overview of how we have made use of open source tools is popular, including Bioconductor, Rmarkdown, version control git, R, and in particular system R package combined with new tools DataPackageR, to apply mild reproducible research workflow for preprocessing of data sets great, perfect for sharing among teams of small-to-medium sized computational scientists.
Our main contribution is DataPackageR tool, which decouples the time-consuming data processing of the data analysis while leaving the track record of how the raw data is processed into analysis-ready data sets. The data object ensure software packages are documented and performs a checksum verification along with the basic package version management, and importantly, leaves record data processing code in the form of sketches package. Our group has been implementing this workflow to manage, analyze and report data pre-clinical immunology test of the multi-center, multi-assay studies for the last three years.