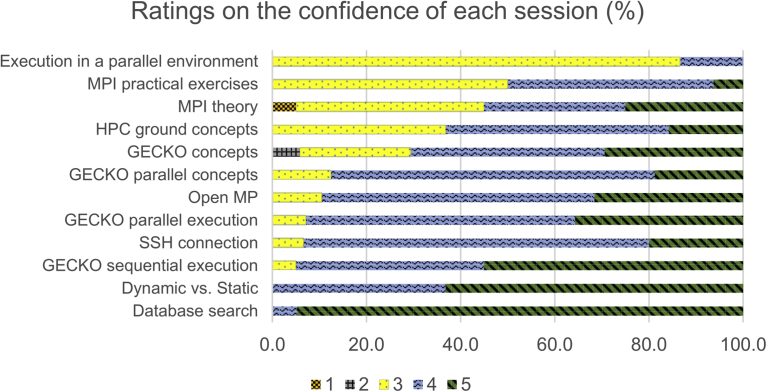
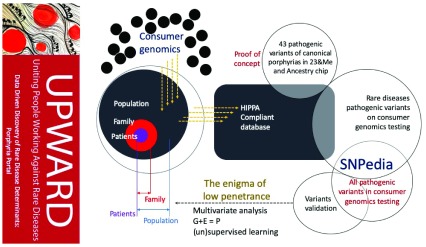
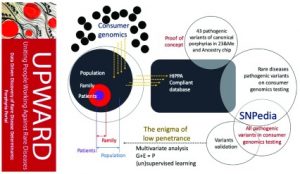
Bayesian network (BN) is a reconstruction of biological systems analysis approach prototype data that has been successfully used to reverse engineer and network models that reflect the various layers of biological organization (from genetics to epigenetics to the cellular pathway for metabolomics).
This is particularly relevant in the context of modern studies (current and prospective), which produces a high-throughput omics heterogeneous datasets. However, there are both barriers theoretical and practical applications for a seamless modeling BN large data such as, including inefficiency optimal computing BN search algorithm structure, ambiguity in the discretization of data, mixing data types, imputation and validation, and, in general, limited scalability in both reconstruction and BNS visualization.
To overcome these and other obstacles, we BNOmics this, improved algorithms and software toolkit to summarize and analyze BNS of omics datasets. Data exploration BNOmics goal in the type of comprehensive biological systems, including both produce new biological hypotheses and test and validate existing ones. Novel aspects of the algorithm centers around improving scalability and application to various types of data (assuming a different distribution of explicit and implicit) within the framework of the same analysis.
Output and visualization interfaces to many available software graphics rendering are also included. Three detailed a variety of applications. BNOmics originally developed in the context of genetic epidemiology data and are continuously optimized to follow the increasing influx of large scale omics datasets available.
Thus, scalability of the software and usability at less than computer hardware exotic is a priority, as well as the application of algorithms and software for the dataset heterogeneous containing polymorphisms many data types-single-nucleotide and other genetic / epigenetic / transcriptome is variable, metabolite levels, variable epidemiology, endpoints, and phenotype, etc.

Predicting gene regulatory networks by combining spatial and temporal gene expression data in Arabidopsis root stem cells.
Identifying transcription factors (TF) and related network involved in the regulation of stem cells is important to understand the initiation and growth of plant tissues and organs. Although many TF been shown to have a role in stem cells Arabidopsis root, a comprehensive view of the signature transcription of stem cells is lacking. In this work, we use data transcriptomic to predict the spatial and temporal interactions between genes involved in the regulation of stem cells.
To achieve this, we are transcriptionally profiled some stem cell populations and develop gene regulatory network inference algorithms that combine with dynamic grouping Bayesian network inference. We utilize our network topology regulator potential major conclusions.
In particular, through mathematical modeling and experimental validation, we identified PERIANTHIA (PAN) as an important molecular regulator of the central functions of silence. The results presented in this work show our combination of molecular biology, computational biology, and mathematical modeling is an efficient approach to identify factors candidate functions in stem cells.